Enjoyed this great NYT article on APOL1, its association with kidney disease in African Americans and the related drug development efforts. It's story telling time. Let's dive into the story of APOL1 from discovery to therapeutics. 🧵
nytimes.com/2022/05/17/health/kidney-disease-black-americans.html
African Americans in US are disproportionately affected by kidney disease. This was first realized almost half a century ago. Here is a 1982 NEJM article succinctly describing when the racial differences in kidney disease first became apparent.
pubmed.ncbi.nlm.nih.gov/7040967/
Today we know individuals of African ancestries are disproportionately affected by kidney disease because they carry certain genetic variants in APOL1 which make them more susceptible to kidney disease *when encountering an environmental trigger such as a viral infection*
It's important to note that APOL1 mutation (which is only the first hit) is alone not sufficient to cause kidney disease. A second hit, typically a viral infection that triggers the interferon signaling is necessary to cause kidney damage.
The APOL1 in kidney disease in Africans is like APOE in Alzheimer's. It's a routine today for genetic studies of kidney disease involving Africans to perform analysis after stratifying the individuals based on APOL1 variants as they alone are a huge source of variation.
But how scientists linked to APOL1 to kidney disease? Why APOL1 risk variants are seen exclusively in individuals of African ancestries? Behind these questions lies a fascinating story of evolutionary arms race between humans and a parasite species called Trypanosome brucei.
APOL1 and trypanosomiasis.
Trypanosomiasis is a vector borne parasitic infection (transmitted by tsetse flies) of the central nervous system and is endemic to regions of Africa. It can be years before the first symptoms appear, which is typically followed by a rapid progression.
When left untreated, the parasites cross the blood brain barrier and infect the brain causing neuropsychiatric symptoms including hallucinations, delirium, mania and the characteristic reversed sleep/wake cycle (hence the name sleeping sickness).
Trypanosomiasis is mainly an animal disease. Humans are naturally immune. Human blood is toxic to the trypanosomes. The parasites explode when they enter the human blood stream.
But over the period of evolution, certain sub species developed protective mutations that enabled them survive in the human blood. The gene (SRA) underlying this protection was identified in 1990s.
However, the magical molecule in the human serum that slaughtered the parasites were not known until 2003. Till then it was referred to as "trypansome lytic factor".
In 2003, through an elegant set of experiments, Vanhamme et al discovered that the trypanosome lytic factor is APOL1, an HDL associated protein.
nature.com/articles/nature01461
The parasites after entering the human blood stream engulf the HDL particles along with APOL1 into their lysosomes. Without SRA (a mutated lysosomal protein), this results in instant death of the parasites.
But in the presence of SRA, the parasites survive. The SRA fights the APOL1 and saves the parasite. The picture shows a model of protein-protein interaction between SRA and APOL1 like two soldiers fighting each other to save their masters.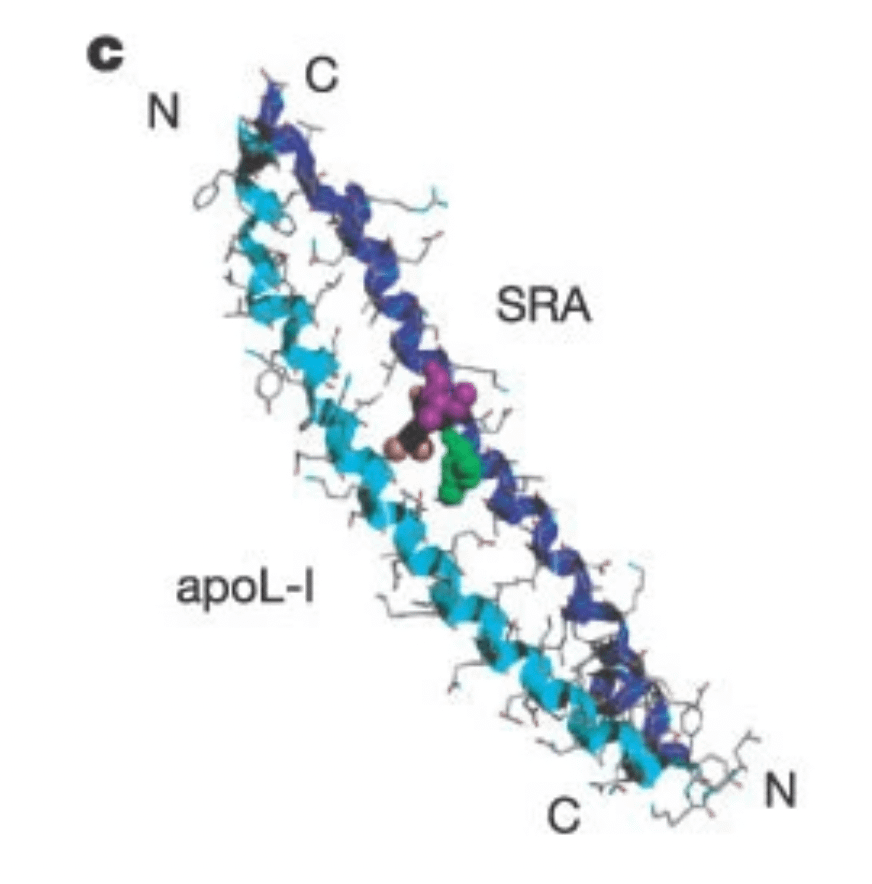
Evolution doesn't take sides. The same evolution that gave the parasites the SRA mutation went on to give humans in Africa protective APOL1 mutations to overcome the SRA and kill trypansomes. But the protection came at a huge price, which brings us to second part of the story.
APOL1 and kidney disease.
The difference in the prevalence of kidney disease between black and white Americans was so big to be fully explained by environmental factors. This motivated the search for genetic explanations.
In 2008, two groups of researchers independently reported in Nature that a genetic locus in chromosome 22 in African Americans strongly associated with non-diabetic kidney disease.
nature.com/articles/ng1008-1145
The authors used a statistical technique called Mapping by admixture linkage disequilibrium (MALD) which works on the prior that the disease associated variants in the admixed African Americans are in genomic regions derived from African ancestors. (Chr 22 peak)
At the time, the scientists believed that the causal gene was MYH9. They didn't know that the real causal gene was actually APOL1. It is fascinating to read these articles today knowing that these signals are driven by APOL1.
Just 2 years later, Martin Pollak (who wrote the commentary of the two Nature articles) and colleagues published in Science that the causal gene at the chr 22 locus associated with kidney disease is APOL1.
science.org/doi/10.1126/science.1193032
Interesting fact: this discovery was made using the sequence data of African individuals of the 1000 genome project (which itself was published only later that year in Nature).
Comparing a mere 205 cases with 180 controls, the authors identified two independent signals at chr 22 locus. The first signal, termed G1, comprised two missense variants in perfect LD and the second a 6 bp deletion termed G2. Both signals replicated in independent cohorts.
The G1 and G2 variants are nearly exclusive to African and African admixed ancestries. An intense positive selection drove these variants to such high frequencies.
With this genetic discovery, the APOL1-kidney disease and APOL1-trypanosomiasis literature began to merge. It turned out the G2 variant was located exactly at the region of APOL1 (C-terminal) that interacted with SRA. How mind blowing is that!
After this discovery, not only the association was replicated numerous times, but a lot of research went into understanding how these mutations exactly rendered APOL1 toxic to kidneys.
Even after decades of research, it's still not exactly clear how APOL1 damages kidney. Except for the fact the damage is almost always initiated by a "second hit" (typically a viral infection like HIV) which spikes the interferon.
nature.com/articles/s41581-022-00538-3
The most fascinating thing that caught my attention in the NYT article is the unfortunate twins who were born with not just with APOL1 variants but also with lupus variants. That's a dangerous combo.
nytimes.com/2022/05/17/health/kidney-disease-black-americans.html
Just few days ago we learnt how an overactive TLR7 caused a severe early onset SLE in a girl. Lupus patients have an overactive immune system. Any viral infection will storm the body with interferons.
twitter.com/doctorveera/status/1520976714200879104?s=20&t=tfMp7zGW5lLJFkgExhFONg
It's not surprising that one of the twins developed kidney disease at an early age of 10 (probably after a viral infection). It's a great example of Gene x Gene x Environment interaction.
Here comes my favorite part.
The APOL1 variants G1 and G2 are gain of function mutations. So the therapeutic approach would be to block APOL1. But one key question needs to be answered: is it safe to inhibit APOL1? The answer came from a farmer in India.
In 2005, doctors at a medical college hospital in Nagpur, India came across an unusual case. A farmer with a type of trypanosomiasis that was never seen before in humans.
pubmed.ncbi.nlm.nih.gov/16172469/
It turned out the farmer was a human knockout for APOL1. Complete lack of APOL1 made him susceptible to even the subspecies (T evansi) that infected only cattle.
nejm.org/doi/full/10.1056/NEJMoa063265
Apart from the infection (from which he fully recovered with treatment) the farmer was apparently healthy. Which was the key piece of information drug developers were desperately looking for.
A similar example from the past.
elifesciences.org/articles/54363
twitter.com/doctorveera/status/1331291385035694082?s=20&t=tfMp7zGW5lLJFkgExhFONg
Thanks for decades of APOL1 research. According to the NYT article, at least 10 companies are currently working on drugs to treat APOL1 kidney disease in Africans and African Americans.
An investigational compound VX-147 (a APOL1 inhibitor) developed by Vertex has already produced positive results in Phase 2. 13 weeks treatment reduced proteinuria by 47% without any major adverse effects.
news.vrtx.com/press-release/vertex-announces-positive-results-phase-2-study-vx-147-apol1-mediated-focal-segmental
Other companies like AstraZeneca and Maze therapeutics are also in the race. Even if one of these drug development efforts become successful, it will a major breakthrough. It might save millions of lives of Africans and African-Americans.
I love the APOL1 story because it's a great example that the scientific research linking racial differences in human health with genetics is not always done with racist motives but sometimes (perhaps often) with the intention of curing diseases and reducing human suffering.

Veera Rajagopal
@doctorveera
🇮🇳 MBBS, MD, 🇩🇰 PhD | 🧬 Scientist @ 🇺🇸 Regeneron | Translating genetic insights into life-saving medicines | Weekly thoughts @ gwasstories.com